
Research
Research on protein dynamics, water networks & molecular allostery.
On my PhD journey to unravel how proteins function and regulate their activities, I focused on a critical yet often overlooked component: water. I developed computational methods showing how hydration shells and internal water molecules influence protein stability, dynamics, and allosteric communication.
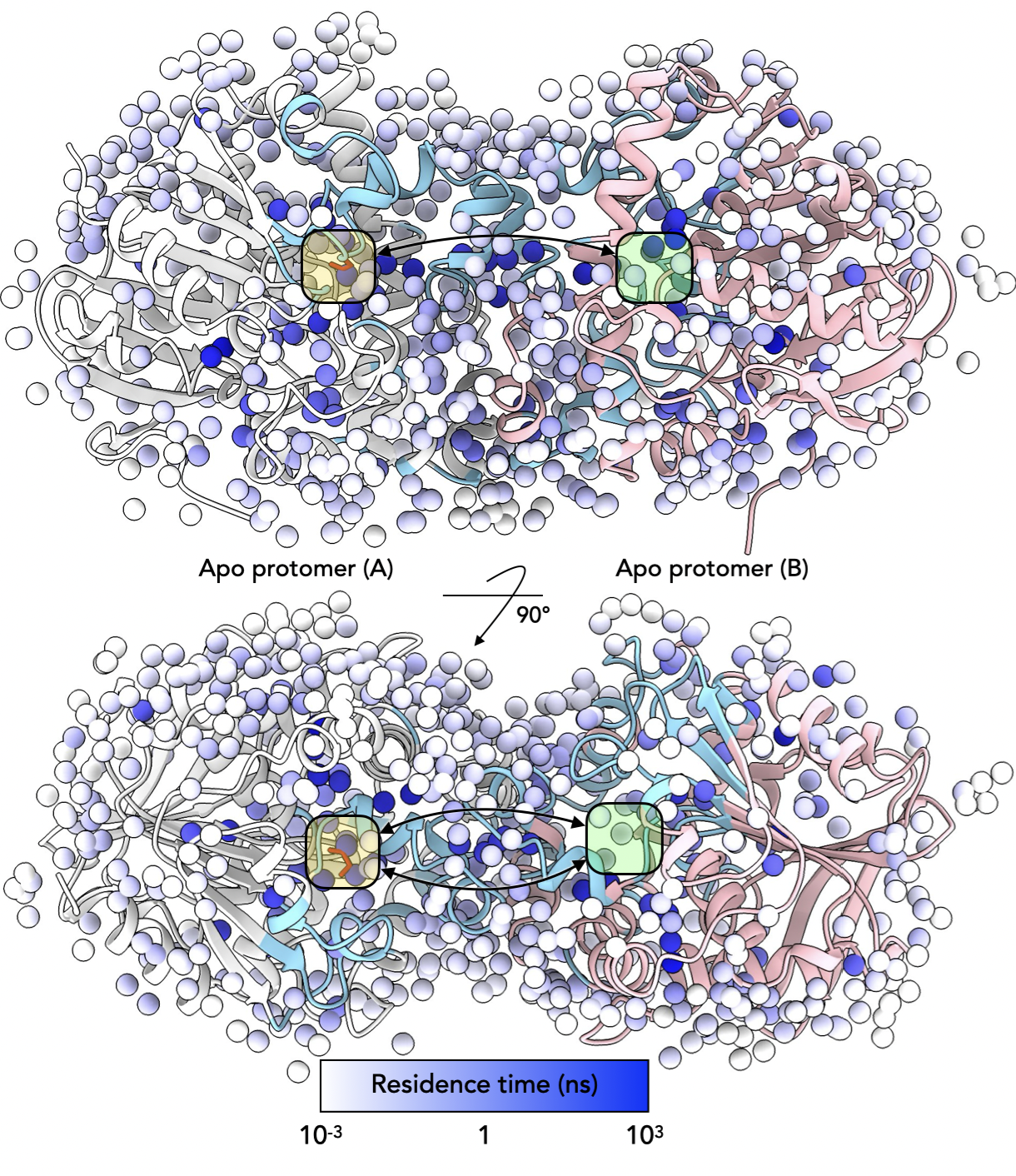
Tracking water–protein interactions
A novel computational approach that accurately tracks how proteins interact with water. By integrating data from X-ray crystallography, I demonstrated the significance of specific water sites that persist in simulations, whether the protein is in a crystal or in solution. This insight generalizes a method for studying how water networks help maintain protein structure and function in native cellular environments.
Crystal vs. solution environments
I explored how the environment — whether a crystal or a solution — affects a protein's conformational landscape. The structural changes observed in a crystal mirror those that occur when a protein interacts with a ligand in solution. This has profound implications for how we interpret time-resolved crystallographic data and understand protein function in its native environment.
Water-mediated allostery
I extended my computational methods to investigate how water influences protein allostery, a process where distant sites within a protein communicate to regulate its activity. My research on a model enzyme revealed that water networks within the protein can stabilize and enhance allosteric signalling when water (H₂O) is replaced with its heavy counterpart (D₂O). This opens up exciting possibilities for protein engineering by manipulating water-mediated communication pathways.
In essence, my research combines molecular simulations with experimental data to draw a complete picture of the vital role water plays in the life of proteins, contributing to a deeper understanding of biological processes at the molecular level.
Methods
Molecular dynamics (MD) simulations
 This approach is sometimes called a "computational microscope"; equations of motion are numerically integrated for every atom based on an empirical potential energy function. Simulations produce long time-resolved trajectories of atomic coordinates and provide a representation of a given molecule's structural ensemble. MD simulations offer a unique advantage over other biophysical techniques by providing an explicit description of the solvent molecules surrounding the protein. This explicit representation of protein-solvent interactions is crucial for understanding solvent-mediated mechanisms of allosteric regulation and functional dynamics. In MD simulations, it is possible to precisely control the protein environment (pressure, temperature, ion concentration, and presence of co-solutes) to mimic experimental or physiological conditions. Comparison of simulations performed under different conditions makes it possible to identify the effects of a wide variety of molecular perturbations. By accurately simulating protein motions and interactions with the surrounding environment, MD simulations can fill in the gaps in our understanding of proteins due to limited experimental data, by providing insights into the free energy landscapes of proteins.
This approach is sometimes called a "computational microscope"; equations of motion are numerically integrated for every atom based on an empirical potential energy function. Simulations produce long time-resolved trajectories of atomic coordinates and provide a representation of a given molecule's structural ensemble. MD simulations offer a unique advantage over other biophysical techniques by providing an explicit description of the solvent molecules surrounding the protein. This explicit representation of protein-solvent interactions is crucial for understanding solvent-mediated mechanisms of allosteric regulation and functional dynamics. In MD simulations, it is possible to precisely control the protein environment (pressure, temperature, ion concentration, and presence of co-solutes) to mimic experimental or physiological conditions. Comparison of simulations performed under different conditions makes it possible to identify the effects of a wide variety of molecular perturbations. By accurately simulating protein motions and interactions with the surrounding environment, MD simulations can fill in the gaps in our understanding of proteins due to limited experimental data, by providing insights into the free energy landscapes of proteins.
Markov State Models (MSMs)
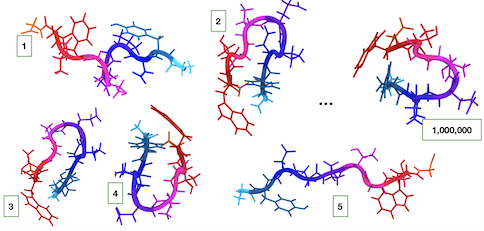 I develop robust, accurate and easy-to-interpret Markov state models for protein dynamics. The steps often include proper featurization of conformational states, extraction of the most significant features through dimensionality reduction, clustering of the conformations into distinct kinetically relevant states, building an MSM transition matrix, and estimating the accuracy of the MSM based on objective quality metrics.
I develop robust, accurate and easy-to-interpret Markov state models for protein dynamics. The steps often include proper featurization of conformational states, extraction of the most significant features through dimensionality reduction, clustering of the conformations into distinct kinetically relevant states, building an MSM transition matrix, and estimating the accuracy of the MSM based on objective quality metrics.
Dimensionality reduction & clustering
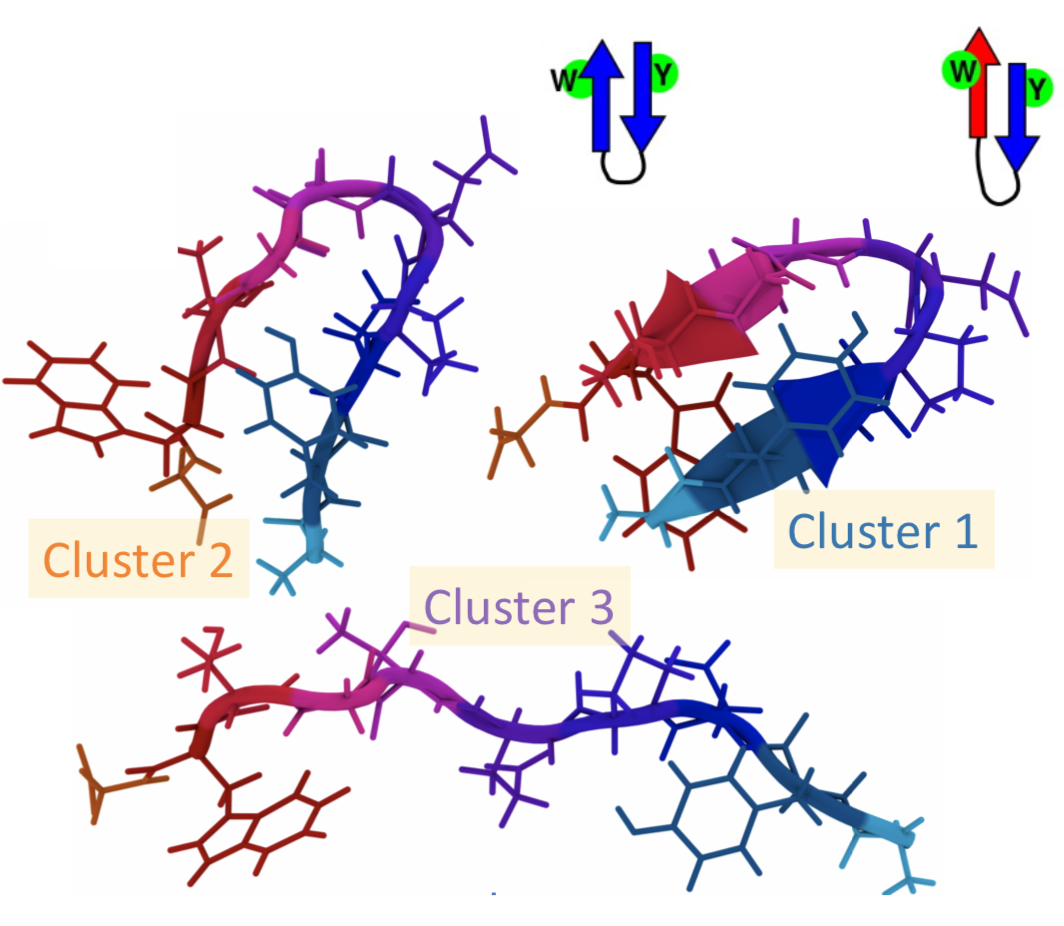
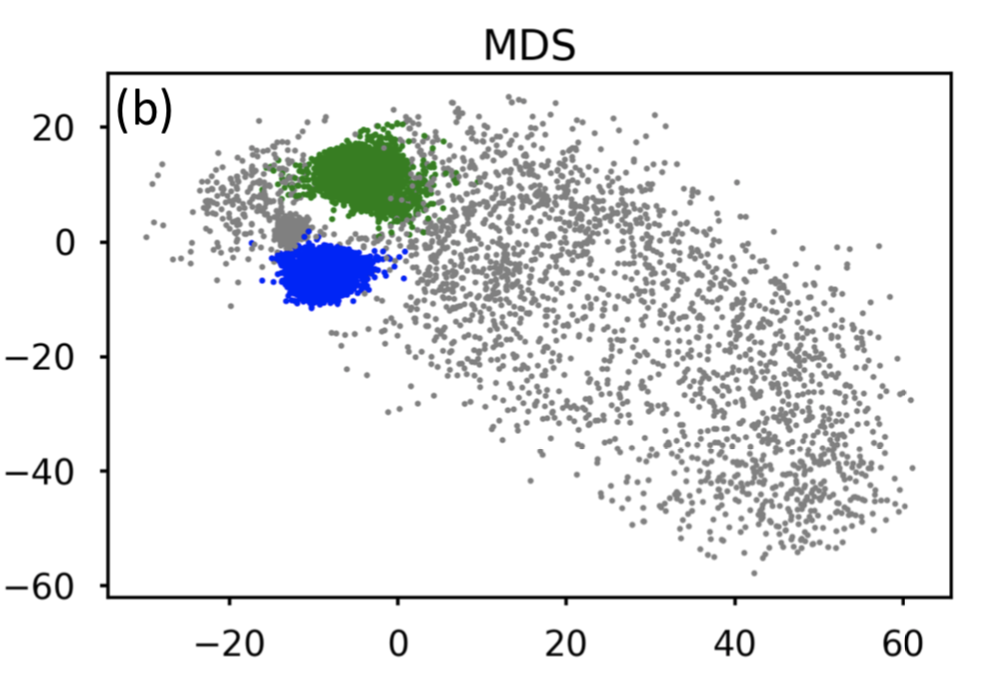 I use dimensionality reduction and clustering of molecular conformations sourced from protein simulations. Clustering methods used to analyze MD trajectories require specification of various parameters, including the number of clusters. The number of clusters is an approximation to the number of conformational states since geometrically and energetically similar structures share the same conformational states. This parameter is often unknown in advance and is the quantity of interest itself, making this problem challenging.
I use dimensionality reduction and clustering of molecular conformations sourced from protein simulations. Clustering methods used to analyze MD trajectories require specification of various parameters, including the number of clusters. The number of clusters is an approximation to the number of conformational states since geometrically and energetically similar structures share the same conformational states. This parameter is often unknown in advance and is the quantity of interest itself, making this problem challenging.